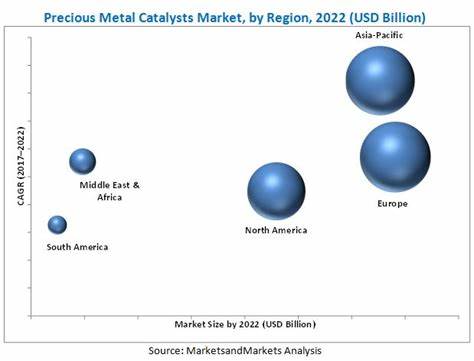
阿尔茨海默病(Alzheimer's Disease,AD)作为全球最常见且最具破坏性的神经退行性疾病之一,困扰着数以百万计的患者及其家庭。尽管传统研究重点聚焦于认知功能下降,但越来越多的证据显示,阿尔茨海默病在发病早期常伴随非认知症状,尤其是嗅觉障碍的出现。嗅觉减退不仅广泛存在于阿尔茨海默病患者中,而且其出现时间往往早于记忆力等认知症状,因而被视为潜在的早期疾病标志。然而,嗅觉障碍背后的具体病理机制尚未明了,限制了早期诊疗手段的发展。最新研究聚焦于脑干内主要的去甲肾上腺能中枢 - - 蓝斑核(locus coeruleus, LC),揭示其轴突在嗅球(olfactory bulb, OB)中的早期退行性改变,是嗅觉功能障碍发病机制的重要环节。蓝斑核作为大脑去甲肾上腺素(noradrenaline, NA)释放的主要来源,其广泛投射调节包括觉醒、注意力及感觉加工在内的多种生理功能。
其中,去甲肾上腺素对嗅觉信息的精细调控尤为关键。通过基因修饰小鼠模型(AppNL-G-F)研究发现,蓝斑核的去甲肾上腺能轴突在嗅球区域出现了显著早期退化现象,时间点甚至早于脑内淀粉样斑块的形成。这种轴突丢失与嗅觉检测能力的降低高度相关,表明去甲肾上腺能系统的退损是促进早期嗅觉障碍的关键因素。值得注意的是,这一轴突损失具有区域特异性,主要局限于嗅球,而非更广泛的蓝斑核投射区域,例如海马、嗅觉皮层或前额叶皮层,说明嗅觉路径尤为敏感。进一步的实验表明,嗅球内的微胶质细胞作为中枢神经系统固有免疫细胞,承担着对轴突残骸的识别和清除作用。在病理状态下,蓝斑核轴突表面异常暴露的磷脂酰丝氨酸(phosphatidylserine,PS)成为微胶质细胞吞噬的"吃我"信号,促使微胶质细胞介导轴突的吞噬。
在体外吞噬能力和基因表达水平的研究中,APP突变小鼠嗅球内微胶质细胞显示出显著增强的吞噬活性,尤其针对去甲肾上腺能轴突,反映出早期的免疫反应与神经退行过程紧密相连。实验中,缺失转位蛋白18kDa(TSPO)的APP突变小鼠表现出抑制微胶质细胞吞噬功能,蓝斑核轴突退化明显减少,嗅觉功能得到部分保护,进一步支持了微胶质细胞吞噬作为轴突丢失机制的核心地位。神经递质去甲肾上腺素的释放受轴突完整性直接影响,通过使用纳去极化敏感探针和双光子显微技术,研究人员证明APP突变小鼠嗅球对气味刺激诱发的去甲肾上腺素释放显著降低。这不仅解释了嗅觉功能减退的生理基础,也凸显了神经元结构完整性对神经递质功能发挥的决定作用。在蓝斑核神经元的电生理记录中发现,这些神经元表现出自发性过度活跃,伴有钙依赖性磷脂酰丝氨酸外翻现象。高频放电导致钙离子内流活化膜脂重组酶,驱动PS外翻,形成微胶质细胞识别的靶标。
超活跃导致的PS暴露成为去甲肾上腺能轴突被过度清除的分子基础,从而加剧嗅觉神经回路的损伤。针对蓝斑核特异表达APPNL-G-F点突变的病毒载体注射模型,研究表明轴突退化及嗅觉缺陷可以独立于其他脑区的变化而产生,进一步验证了蓝斑核去甲肾上腺能系统的核心作用。人类临床及病理学研究同样支持这一观点。早期阿尔茨海默病患者的嗅球组织中蓝斑核去甲肾上腺能纤维密度显著下降,且通过转位蛋白18kDa(TSPO)PET成像发现,嗅球区域存在微胶质细胞数量增加或激活状态提升,这些变化同样与嗅觉识别能力减退相伴。值得注意的是,这种微胶质细胞密度的增多主要反映细胞数量增加而非激活强度的改变,提示微环境免疫细胞扩增在早期疾病阶段的特点。嗅觉异常不仅是早期临床表现,也被视为预示认知功能进一步下降的预警指标。
结合蓝斑核-嗅球通路的退化机制,嗅觉功能检测联合神经影像学手段有望成为早期诊断阿尔茨海默病的重要工具。未来的研究将进一步明晰该系统介导的疾病进展路径,探讨针对微胶质细胞吞噬活性调控以及保护蓝斑核去甲肾上腺能轴突的干预方法,或将推动开发延缓或阻止阿尔茨海默病发展早期阶段的创新治疗策略。综合来看,阿尔茨海默病早期蓝斑核去甲肾上腺能轴突的选择性退化,尤其是嗅球区域的微胶质细胞介导的吞噬机制,是嗅觉障碍发生的基础。轴突损失导致的去甲肾上腺素释放减少,进而影响嗅觉信息处理,构成了疾患认知症状显现前的重要神经病理改变。锁定该过程为研究重点,不仅有助于揭示阿尔茨海默病早期发病机制,还为临床早期诊断和个性化治疗提供了科学依据和潜在靶点,具有重要的理论与应用价值。 。