计算化学是研究分子和材料性质的关键科学手段,广泛应用于药物设计、材料开发及能源技术等领域。然而,长期以来其预测准确性受限,阻碍了真正依赖计算模拟替代耗时繁琐实验的进程。近年来,深度学习技术的飞速发展为计算化学带来革命性契机,尤其是在提升密度泛函理论(DFT)准确性方面取得了突破性进展。此次成果不仅大幅缩小了DFT与实验结果之间的误差,还为分子设计和材料科学注入了强劲动力,推动科学发现步入全新纪元。密度泛函理论作为计算化学的主力方法,凭借较低的计算复杂度和较快的模拟速度,成为研究原子和分子电子结构的基础工具。由诺贝尔奖得主Walter Kohn提出的DFT,将复杂的多电子薛定谔方程重构为电子密度的函数,极大提高了计算效率。
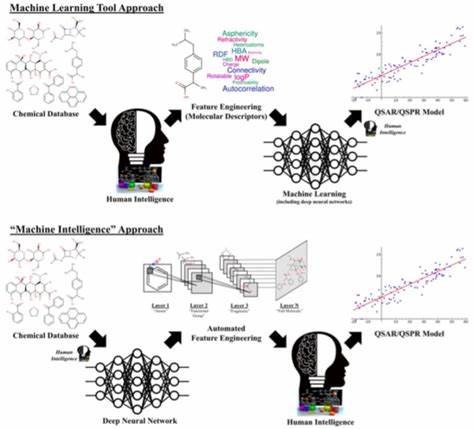
然而,DFT的核心难题在于交换-相关(XC)泛函的精确表达尚未被揭示,迄今为止研究者依赖经验和数学物理的近似表达,导致预测误差难以满足高精度科学研究的要求。大多数传统XC泛函的误差通常远高于化学精度的1 kcal/mol限制,使其更多地用于解释实验结果,而非可靠预测新分子的性质。深度学习的优势在于能够从大量高质量数据中自动学习复杂的非线性关系,且具有强大的泛化能力。微软研究院与合作团队突破了以往机器学习尝试仅限于传统手工设计特征的局限,采用端到端的深度神经网络直接处理电子密度,重塑了XC泛函的表达形式。该模型命名为Skala,通过学习海量、极为准确的高精度波函数计算数据,成功实现了前所未有的预测精度,误差显著缩小至接近实验水平。为了训练这样强大的模型,科学家首先面临数据瓶颈。
高精度的波函数方法虽能生成接近真实物理的电子能量数据,但计算成本极高。团队联合澳大利亚新英格兰大学的权威专家,构建了涵盖丰富分子结构的大型高质量训练数据集,规模远超以往研究。借助微软Azure强大算力支持,完成了对数十万分子结合能数据的精准计算,为深度学习模型训练奠定坚实基础。这些数据涵盖了多种分子类型,确保模型能够学会捕捉电子密度与能量之间复杂的对应关系,并在未知分子上能够准确预测其化学性质。Skala模型不仅突破了传统DFT的计算瓶颈,在保持计算资源经济性的同时,实现了增强的预测能力,还在多个公认基准测试上超越了现有最佳XC泛函,尤其是在W4-17和GMTKN55数据集中的表现堪称革命。模型在单参照体系的原子结合能预测中达到了平均误差低于0.85 kcal/mol的质量水平,首次使DFT预测能够被科学家们作为实验结果的可靠替代。
该成果具有里程碑意义,为科学团队将来加速新药分子筛选、高性能电池材料设计及环境友好型肥料开发提供了强有力的计算支持。深度学习模式的引入不仅提升了理论模拟的准确性与效率,更打开了前所未有的可能性,例如针对更大、更复杂分子体系的扩展,或是将准确度提升至更广泛的材料科学问题,如催化剂设计和碳捕获技术。微软也推动建立了DFT研究早期访问计划,邀请产业和学术界共享最新XC泛函和相关数据资源,加快成果转化并推动跨界合作。业内公司如默克和Flagship Pioneering等均对该技术高度评价,坚信深度学习辅助的DFT将成为推进数字化化学的重要利器,为工业创新带来革命性变革。从航空工程模拟到计算化学的转型类比,深度学习不仅缩短了模拟周期,更实现了设计方案的精准预测。预计未来,结合现代计算平台和大规模高精度数据的能力,将使得基于DFT方法的计算模拟成为如航空设计般成熟可靠的科研日常工具,极大降低实验成本和时间,助力科学家更快实现理想分子和材料的发现。
总的来看,深度学习赋能的密度泛函理论成功解决了数十年来困扰计算化学领域的根本难题,为科学研究和应用开发提供了新的范式。借助不断扩充的准确数据集和优化的神经网络架构,Skala等模型展现出极高的潜力。未来的研究将更加聚焦于模型对更广阔化学空间的适应性,提升异质材料体系的预测能力,及结合量子计算等前沿技术,实现从根本上多尺度、多物理过程的精准建模。此场科研突破预示着计算化学进入一个由智能算法驱动的黄金时代,促进绿色能源、生命科学、新材料等多个重大领域的科学进展,推动工业界向全模拟化设计发展目标迈进。随着更多开放资源和协作平台的问世,全球科学家共同努力,将让深度学习推动的计算化学技术惠及更广泛的创新应用,助力人类文明迈向更绿色、更智能的未来。