结构基础药物设计(Structure-Based Drug Design,SBDD)作为现代制药研发的基石,依赖于高分辨率三维蛋白质结构信息指导药物分子的优化设计。传统上X射线晶体学因其解析精度高、应用广泛而成为结构获取的主流手段。然而,晶体学在蛋白质结晶难度大、动态信息缺失以及无法直接观测氢原子等方面存在不可忽视的局限性,促使科学家寻求新的补充或替代技术。核磁共振(NMR)技术凭借原子级别的分辨率和能够在溶液状态下捕获分子动态的优势,正在引领结构基础药物发现迎来新的发展机遇。核磁共振驱动的结构基础药物发现(NMR-SBDD)方法通过揭示蛋白质与配体的分子相互作用,不仅克服了传统技术难以解决的问题,还为药物研发提供了更加丰富和动态的结构信息。理解蛋白质-配体间非共价作用的本质,如氢键、盐桥、π-相互作用及疏水力学,能够直接指导药物分子的精细修饰,提升结合亲和力与选择性。
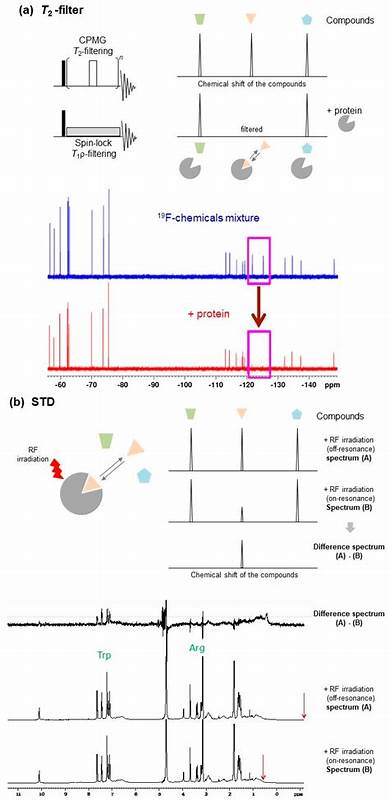
NMR技术通过选择性氨基酸侧链标记和先进的核磁谱仪硬件,加上计算机辅助的数据分析,将蛋白质-配体复合物的信息提炼为详实的结构和动力学数据。与依赖静态晶体结构不同,NMR可以描述蛋白质和配体的柔性变化,捕捉其存在的多种结合构象及相互转换过程,这对药物设计中合理评估结合热力学和动力学参数至关重要。另外,NMR独具探测氢原子的能力,揭露传统晶体学无法直接观察的重要氢键及水合层,这对于精确理解结合界面和稳定配体结合位点的水分子网络格外关键。水分子在蛋白质-配体界面扮演桥接和调节角色,NMR技术能够探测不同寿命和运动状态的水分子,为水模拟配基的设计提供实验依据,从而实现结合能的优化。NMR技术在弱相互作用和低亲和力配体筛选,尤其是片段一体化药物发现(Fragment-Based Drug Discovery,FBDD)中展现出极大潜力。其高灵敏度和动态检测能力使得科学家能识别短暂、微弱却关键的CH-π、甲基-π等相互作用,及时捕捉早期片段的结合模式,有助于后续分子的结构优化。
借助碳-13氨基酸前体的定点标记,NMR谱图显著简化,提升大分子或复杂体系的信号解析效率,同时配合人工智能驱动的信号处理,极大加快谱图分配与信息提取速度。近年来,计算生物学方法如分子动力学模拟和深度学习算法与NMR数据的结合为构建真实存在的蛋白配体构象全集成为可能。AlphaFold等先进结构预测工具虽能生成多个蛋白构象,但缺乏实验验证的动态信息。通过将NMR化学位移扰动(CSP)作为约束,科学家可对多样化计算结构进行重新加权,筛选出最符合溶液状态实际情况的结构,建立动态、准确的复合物结构集。这种融合计算与实验的策略极大提高了结构预测的活用价值,为药物设计中的机制研究及诱导契合与构象选择模型提供有力支持。此外,NMR还能深入揭示蛋白质-配体复合物的动力学特征。
诸如侧链翻转、骨架局部运动及构象转换等微妙的动态过程均可用先进NMR实验技术捕获,帮助药物设计者理解结合的热力学补偿、结合速率及复合物稳定性。这为突破传统静态结构限制,设计更高效且持久的候选分子奠定基础。尽管如此,NMR技术在蛋白质分子量较大及高通量筛选方面仍面临挑战。分子量增加导致谱图重叠、信号灵敏度下降,限制了应用范围。近年来,诸如TROSY系列技术、新型标记方法及动态核极化等硬件突破为解决此类瓶颈贡献力量。与此同时,通过机器学习推动的自动化数据分析流畅整合实验及计算输入,未来有望实现NMR在药物筛选中的大规模应用。
NMR驱动的结构基础药物设计不仅能够加速早期筛选命中的结构鉴定和优化,还适合处理传统晶体学难以解决的复杂系统,如含有柔性连接区、固有无序区的蛋白,也对研究生物大分子凝聚体提供新视角。展望未来,随着硬件技术进步、AI与机器学习的深度融合,NMR实验和计算模型的结合将更趋完善,为动态结构群体的解析、隐匿口袋识别和全谱结合机制阐释带来革命性突破。多模态结构生物学整合多种技术优势,结合NMR的原子级别动态细节,有望实现对靶标分子极致精准的结构和功能解析,极大推动靶向药物开发的创新和成功率。在分子设计层面,深入理解和利用分子间动态非共价相互作用、结合界面水分子网络及构象多样性,有望帮助开发出新型高效、选择性强的精准治疗药物,覆盖传统难以攻克的靶标及疾病领域。综上所述,NMR驱动的结构基础药物发现正成为突破经典药物设计局限的关键力量。它通过揭示蛋白质与配体之间丰富的分子相互作用和动态行为,为制药科学提供了前所未有的洞察视角和工具平台。
持续优化实验技术、宣传多学科融合及提升高通量处理能力,将进一步放大其在药物研发中的作用和价值。随着相关技术和理论体系的成熟,期待NMR-SBDD引领全球药物发现步入精准高效的新纪元,为人类健康福祉创造更多可能。