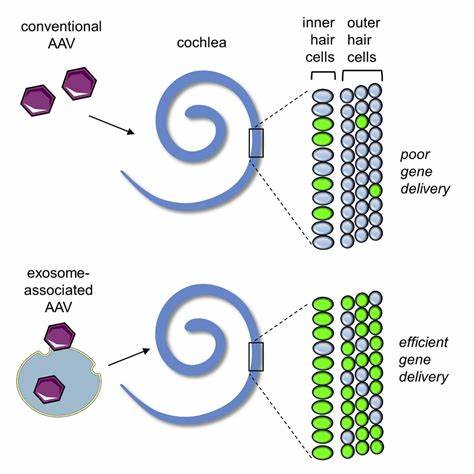
耳聋作为全球最常见的感官障碍,具有极其复杂的遗传背景。迄今为止,已有超过150个基因与遗传性听力障碍相关,其中CLIC5基因因其突变引发的DFNB103型遗传性聋哑成为科学界关注的焦点之一。CLIC5编码氯离子通道蛋白,对内耳毛细胞的结构稳定性及功能维持起着关键作用。缺失或突变导致毛细胞的纤毛结构异常,进而造成听力和前庭功能的损害。面对传统治疗手段难以根治遗传性耳聋的困境,AAV介导的基因疗法为靶向修复基因缺陷提供了新的突破口。AAV作为一种非致病性病毒载体,具有安全性高、转导效率优良的优势,广泛用于神经系统及感觉系统的基因递送。
研究者利用两种不同形式的AAV载体 - - 单链AAV(ssAAV)和自补链AAV(scAAV)来输送野生型CLIC5基因至缺陷小鼠耳内,取得了显著的疗效提升。自补链AAV因其双链结构,可绕过宿主细胞第二链DNA合成的速率限制,快速启动转基因表达,表现出更高的表达效率和更低的给药剂量需求,这对于降低潜在的免疫反应及毒性意义重大。鼠标模型展现出因CLIC5缺失导致的显著听力下降以及行为异常,如旋转行为和运动障碍,对应人类患者的听力丧失与平衡功能障碍。通过早期(新生日)经由内耳前庭注射AAV载体,科学团队成功恢复了CLIC5蛋白在耳蜗内外毛细胞及前庭毛细胞的表达。治疗不仅阻止了毛细胞纤毛的异常融合与延长,还显著提升了毛细胞的存活率。功能检测如听觉脑干反应(ABR)及耳声发射(DPOAE)结果显示,治疗组小鼠的听觉阈值明显降低,听力功能得到有效修复。
前庭功能的恢复也在开放场地行为测试及加速转杆实验中得到了验证,治疗后小鼠的旋转行为明显减少,运动协调能力提高,平衡功能趋近正常水平。此外,自补链AAV载体以低于单链AAV一个数量级的滴度,达成同等的疗效,极大提升了临床转化的可行性和安全性。科学家们认为,CLIC5相关遗传性耳聋在人类患者中发病时间相对较晚,多在儿童早期至青少年阶段出现听力和前庭功能逐步减退,与模型动物表现的进展性病理极为相似。这一特性为采用基因疗法提供了更宽广的治疗窗口。早期干预可有效阻止毛细胞退行性变化,预防听力丧失的不可逆损伤。研究中还强调了使用特异性启动子和优化病毒载体的必要性。
通过精选表达元件,如鸡β-肌动蛋白衍生启动子(CB6)及优化载体壳,保障基因表达强度和稳定性,实现精准调控和高效治疗。基因疗法方案的成功同时对未来多基因、复杂遗传背景的耳聋治疗提供了宝贵借鉴。当前,AAV载体在体积容量、免疫原性以及大规模生产中仍面临挑战。特别是自补链AAV因基因组容量限制(约2.4kb),只适用于体积较小的基因。但值得庆幸的是,包含CLIC5在内的大部分听力相关关键基因均符合这一限度,为后续推广应用奠定基础。此外,减少给药剂量不仅降低潜在免疫反应和炎症风险,还有利于生产成本控制及广泛推广。
未来的研究需结合临床试验进一步评估预约给药时机、剂量优化、长期疗效及安全性,以期实现真实患者的治愈或功能大幅改善。AAV介导的CLIC5基因替代治疗不仅彰显了精准医疗对耳聋治疗的变革力量,更象征着听力学领域从症状管理向根本治愈性的深度转变。随着技术革新和临床试验的逐步推进,基因疗法将在未来成为遗传性及获得性耳聋患者的标准治疗选项。与此同时,相关伦理、监管和患者教育方面也需同步加强,确保技术惠及更广大有需求的人群。总结来看,基于AAV的基因治疗为解决CLIC5相关的耳聋与平衡功能障碍提供了切实有效的策略。科学家们利用先进的病毒载体技术成功恢复了内耳毛细胞的功能结构,翻转了小鼠模型的听力和运动缺陷,展现了极高的临床应用潜力。
随着对这一领域理解的深化,未来将有更多个性化、针对性的基因治疗方案持续涌现,推动听力障碍治疗进入崭新纪元。 。