上皮组织维持稳态依赖于细胞的不断更新与清除。细胞分裂与死亡之间的平衡一旦被打破,屏障功能会受损或异常增生就会发生。近年来,关于机械力如何影响上皮细胞命运的研究揭示了一个关键现象:在拥挤的微环境中,活细胞会被挤出并随后死亡,从而维持组织的细胞数恒定。然而,为什么在同一拥挤区域中只有部分细胞被选中挤出?最新研究表明,能量状态与膜电位的差异成为决定性因素,离子通道与水通道在这一选择过程中发挥核心作用,形成了一套从感受拥挤到执行挤出的完整机制。 在传统认知里,Piezo1等应力敏感离子通道因其对拉伸的响应而被视作上皮细胞对机械刺激的首要探测器。Piezo1的激活可诱导细胞分裂或挤出,这一通路随后通过S1P信号介导Rho依赖的肌球蛋白收缩来完成细胞的顶向挤出。
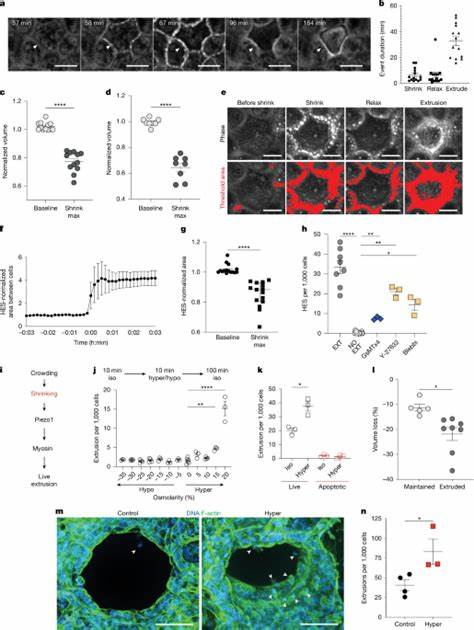
然而,观察到的事实是,许多挤出事件并未伴随明显的局部钙信号上升,也没有显著的细胞干质量下降。相反,研究人员发现多数被挤出的活细胞在挤出前都会经历短暂但显著的体积丧失,即通过水外流引起的细胞收缩。这种初始的体积丧失既不同于经典的凋亡性体积减少,也并不依赖肌球蛋白驱动的收缩。更重要的是,这一过程是由膜电位变化和特定离子通道的激活所介导的。 在分子层面上,研究指出上皮钠通道ENaC在拥挤刺激下可被激活并允许钠离子进入细胞。ENaC位于细胞顶端,具有机械敏感性,当细胞因拥挤产生局部应力时,ENaC通道会开启,带来钠离子内流并引发膜去极化。
对于大多数活细胞而言,细胞内钠浓度的上升能被Na+/K+ ATP酶迅速纠正,泵出钠并恢复膜电位。但那些细胞能量储备不足、ATP含量较低时,Na+/K+ ATP酶无法高效工作,导致细胞持续去极化。膜去极化为电压门控钾通道Kv1.1和Kv1.2提供触发条件,随后这些钾通道开放致使钾离子外流,并与体积调节相关的氯通道如SWELL1协作促成水分外流,从而导致细胞快速收缩。当收缩幅度超过一定阈值(实验中大约为体积的17%)时,细胞便进入挤出通路,并最终被邻近细胞通过S1P及Rho介导的皮质收缩所顶出。 这种以能量状态为筛选标准的模式具有逻辑经济性。上皮细胞要维持紧密的屏障与集体功能,必须优先保留那些具备足够代谢储备以应对环境变化和履行生理任务的细胞。
ENaC作为一个"压力传感器"并非只是单纯检测机械拉伸,而更像是在拥挤条件下对细胞电生理稳定性的探针。它通过短时诱导钠入流来"试探"细胞是否有足够的能量去维持膜电位。如果能量充足,细胞通过ATP驱动的离子泵恢复稳态;如果能量不足,细胞将持续处于去极化状态并被动地通过电压门控离子通道引发体积丧失与挤出。 实验证据支持这一链路的几个关键环节。首先,使用染料与遗传编码传感器同时监测细胞ATP、膜电位和离子浓度的时间序列显示,ATP水平在去极化之前下降,钠内流紧随其后,膜去极化发生在收缩之前。其次,药理学干预验证了各离子通道的功能定位:阻断ENaC能显著降低去极化和后续收缩,抑制Kv1.1/Kv1.2或SWELL1可干预体积丧失并抑制细胞挤出。
此外,通过人为制造的短期高渗处理来诱发细胞脱水,也能提升挤出速率,说明体积阈值在触发挤出中至关重要。再者,减少细胞ATP(例如应用线粒体ATP合酶抑制剂)会增加收缩与挤出的发生,补充底物如葡萄糖则可以降低挤出率,进一步验证了能量在决定细胞命运中的核心作用。 从组织尺度看,这一机制帮助解释为何挤出倾向于发生在拥挤区域而非随机分布。细胞在拥挤中不断相互挤压,ENaC在多个细胞中被短时激活,但只有相对能量不足的个体无法借助Na+/K+ ATP酶恢复膜电位,才会进入去极化 - 钾离子外流 - 水外流的链式反应,最终被挤出。这样的选择性避免了在细微且普遍的机械扰动中频繁地误触发Piezo1与后续挤出通路,体现了组织在稳定性与更新间的平衡策略。 这一新认知对疾病理解和临床干预都具有重要启示。
许多慢性疾病,如糖尿病和某些代谢综合征,会导致局部组织的代谢异常与能量供应不足。在这些背景下,上皮细胞更易被能量缺乏所"标记"并被持续性挤出,可能造成屏障功能长期损害。呼吸道疾病中的气道上皮、肾脏中的皮质上皮以及肠道上皮都高度依赖稳态挤出与更新,若能量相关的选择机制被扰乱,可能加剧组织脆弱性或促发慢性病变。此外,肿瘤学视角指出,癌细胞常常通过代谢重编程逃避凋亡与清除。如果肿瘤细胞能保持较强的ATP供应或调节ENaC/Kv通路,它们可能在局部竞争中占据优势,从而绕过以能量为主的淘汰机制。这提示离子通道与代谢通路可能成为调控细胞竞争与组织稳态的潜在治疗靶点。
在药物与治疗策略上,对ENaC、Kv通道或SWELL1的调节具有双面效果。抑制ENaC可以减少拥挤触发的钠入流与后续去极化,从而降低细胞被选择性挤出的概率,这在某些需要保护脆弱上皮的急性期或修复期可能有益。相反,在需要促进有害细胞离开的情景下,例如清除受损或感染的细胞,适当增强ENaC或下游体积调节通路的响应可能有助于加速清除。对于癌症治疗而言,若能有选择性地干预能量代谢或膜电位差,使肿瘤细胞失去维持膜电位的能力,就可能诱导其被周围正常细胞驱逐,但这类策略需要极高的选择性以免伤及健康组织。 基础研究层面还存在若干关键问题值得深入探索。ENaC为何对拥挤的力学特征敏感,其结构或调控元素如何被机械微环境所影响,仍需更细致的分子与生化解析。
Kv1.1与Kv1.2在上皮细胞的分布、与紧密连接蛋白及水通道的空间协同如何实现,也需要进一步的成像与分子互作研究。此外,Piezo1在本机制中的下游角色值得厘清:细胞收缩如何触发Piezo1介导的钙信号并启动S1P释放,乃至完成邻居细胞的收缩与挤出执行,仍是连接早期电生理事件与最终机械行为的关键环节。 实验方法学的改进同样推动了发现进程。量化相位成像用以测量干质量变化、基于膜电位与ATP的荧光蛋白传感器、以及"lightning"类的半自动化图像分析工具都加速了对短时体积变化和离子动态的捕捉。动物组织切片的外周精切片模型(PCLS)为在更接近生理的环境中验证体外发现提供了桥梁,是评估在器官尺度上该机制是否普遍存在的重要平台。 总体来看,拥挤诱导的上皮细胞挤出并非单一的机械被动事件,而是一套由代谢能量、膜电位与离子通道协同构成的选择性清除机制。
能量匮乏为被动淘汰提供了筛选依据,ENaC充当了拥挤探针,Na+/K+ ATP酶的效能决定了细胞能否渡过短暂的离子扰动,电压门控钾通道与体积调节离子通道则将微小的电生理差异放大为决定性体积丧失与挤出行为。理解并利用这一链路,将有助于开发调控上皮稳态的新方法,改善在代谢疾病、气道损伤或某些肿瘤微环境中异常细胞清除失衡所带来的病理后果。 未来研究需要整合细胞生物学、电生理学与代谢学的多学科方法,更精细地绘制出上皮细胞在拥挤中的命运地图。与此同时,基于离子通道的小分子或生物制剂的开发应当考虑组织特异性与代谢背景,以期将调控细胞挤出的策略转化为可控且安全的治疗工具。随着我们对能量选择性挤出机制的理解逐步深化,上皮组织如何在拥挤与需求之间实现自我修复与更新的秘密也将被逐步揭示。 。